Skin development, homeostasis, carcinogenesis and regeneration
Via a comparative transcriptome approach, we identified the ∆Np63 isoform of the p63 transcription factor, a close relative of the tumor suppressor p53, as a direct transcriptional target of BMP signaling. Antisense-mediated loss of ∆Np63 functions in zebrafish embryos leads to severe lesions in the embryonic epidermis, most likely due to a precocious differentiation and reduced proliferation of basal keratinocytes. This phenotype is very similar to that of human patients with p63 mutations, indicating that crucial principles in the genetic control of early epidermal development are conserved between fish and mammals (Bakkers et al., Dev. Cell 2002). Encouraged by this finding, forward genetic screens in zebrafish were carried out to identify more and novel regulators of early skin development. In addition, applying reverse genetics technology, loss-of-function mutants in different candidate genes were generated, such as in TAp63, another isoform of p63.
In the forward genetic screens, three different classes of embryonic skin phenotypes were obtained: (A) Skin degeneration, particularly in the median fins; (B) Blistering / compromised epidermal-dermal junction; (C) Epidermal aggregation / loss of epidermal integrity.
Epidermal-dermal junctions
Subsequent identification of the causative mutations in phenotype classes (A) and (B) via meiotic mapping and/or whole genome/exome sequencing revealed that loss of the basement membrane component Laminin alpha5 (Lama5) or its integrin receptor Itga3 on basal keratinocytes leads to epidermal degeneration (A), while embryonic skin blistering (B) is caused by loss of different components of the dermal extracellular matrix, such as Fibrillin-2, the Fraser complex components Fras1, Frem1 or Frem2, or the thus far little understood Fibulin family member Hmcn1 (Carney et al., PLoS Genet. 2010). In human, loss-of-function mutations in Fras1, Frem1 or Frem2 cause Fraser Syndrome, making the corresponding zebrafish mutants to valuable in vivo models for this severe and multi-symptome, but little understood congenital human disorder (see also Research Area “Extracellular matrix and musculoskeletal systems”).
Embryonic epidermis and carcinogenesis
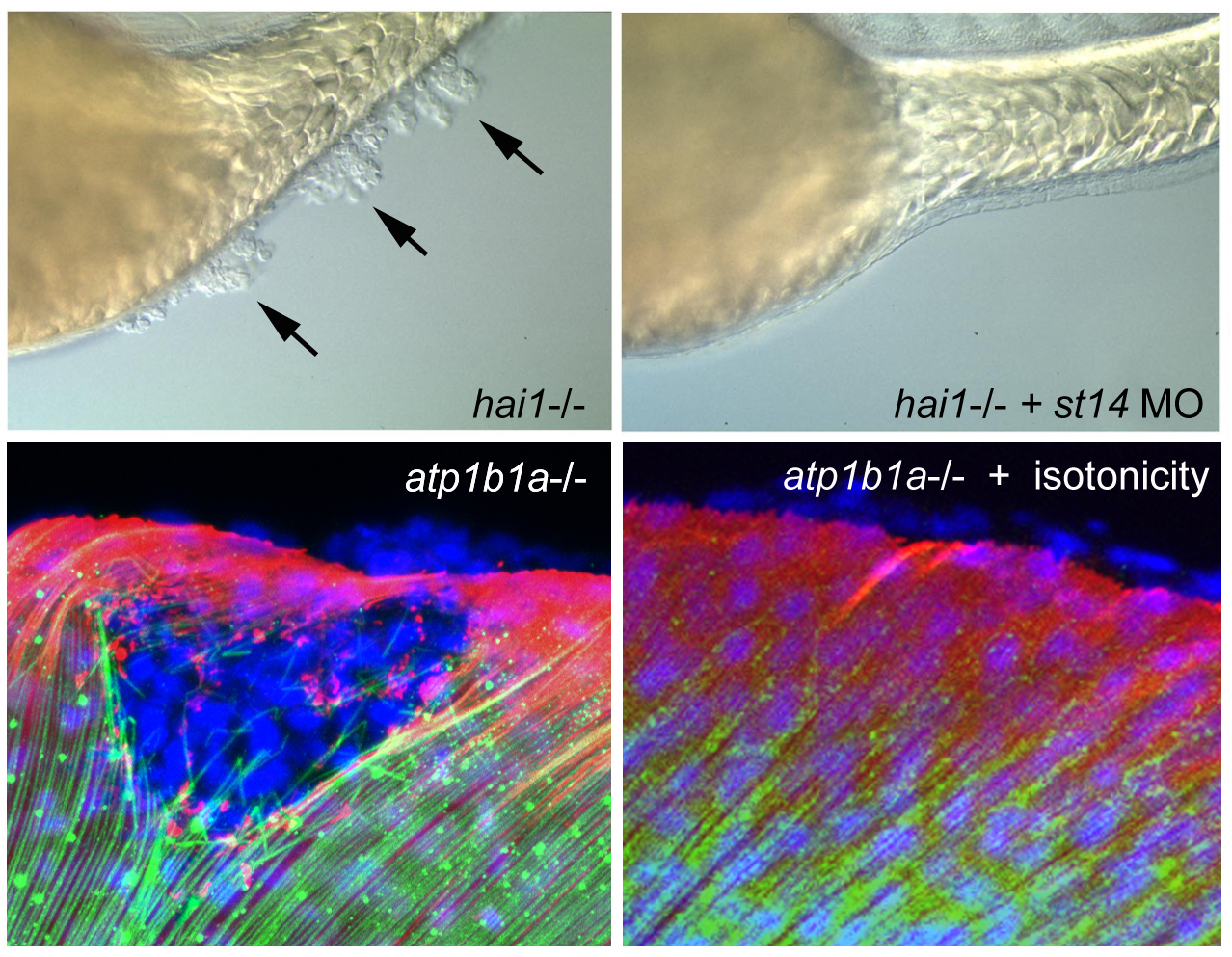
Applying similar gene identification approaches for the epidermal aggregation mutants (3), we could show that the epithelial cell adhesion molecule Epcam (Slanchev et al., PLoS Genet 2009), a clathrin subunit (unpublished), the clathrin-associated protein Clint1 (Dodd et al., Development 2009), the Hepatocyte growth factor activator inhibitor Hai1 (Carney et al., Development 2007), and the Na/K-ATPase beta subunit Atp1b1a (Hatzold et al., eLife 2016) are required for proper epithelial integrity of the embryonic epidermis. The phenotype of the clathrin and clint1 mutants suggests that clathrin-dependent endocytosis and/or recycling of membranous proteins is required for epidermal homeostasis, a notion currently analyzed in more detail.
hai1 mutants display an up-regulated activity of the extracellular protease Matriptase1 (ST14), which in turn causes a loss of epithelial integrity and epidermal hyperproliferation, constituting a model of embryonic carcinogenesis (Carney et al., Development 2007). Interestingly, ST14 displays a dual face, not only promoting epidermal hyperplasia and neoplasm, but also entosis of transformed basal keratinocytes via upper peridermal cells and the apical extrusion / shedding of such cell complexes, constituting a tumor-suppressive mechanism, and accounting for self-healing of weak hai1a alleles. This dual face is achieved via a branching of the ST14 downstream pathway at the level of phospholipase D, leading to the activation of mTORC1, promoting hyperplasia, as well as sphingosin-1-kinase, promoting sphingosine-1-phosphate (S1P) production and thereby entosis and apical cell extrusion as tumor-suppressive processes (Armistead et al., 2020).
Of note, in contrast to the weak hai1a allele, zebrafish mutants with strong / null hai1a allele, although also displaying elevated S1P-mediated apical cell extrusion rates, do not survive (Armistead et al., 2020), but die during early larval stages. According to our unpublished data, this is due to increased death rates of epidermal cells, which is not seen in the viable weak allele and which leads to compromised epidermal integrity and barrier function. We could further show that only the strong, but not the weak, hai1a mutants display increased levels of ceramide (which is a substrate of sphingosin and S1P), and that it is a ceramide-dependent apoptosis by which the epidermal cells of strong hai1a mutants die. But why should increase usage of ceramide for ST14-induced production of S1P should lead to an increase (rather than a descrease) of ceramide levels? Experimental and theoretical modelling approaches showed that this is due to the overshooting of a negative feedback loop from ceramide to the ceramide synthase genes CeS5 and CeS6. In sum, this shows the ceramide – sphingosine rheostat is highly sensitive to the strength of ST14 interference at the level of sphingosin-1-kinase. While moderate interference leads to the activation of apical cell extrusion as a gentle, epithelia-maintaining mechanism of tumor suppression, stronger interference preferentially leads to the induction of ceramide-dependent apoptosis, which is an even more efficient tumor-suppressive process, but less tissue-preserving, so that the animals do not die by cancer, but by the loss of skin barrier function.
More persistent and stronger epidermal malignancy than in hai1 mutants takes place in atp1b1a mutant embryos, in which basal keratinocytes do not only hyper-proliferate, but also degrade the cutaneous basement membrane and invade dermal compartments as well as inner tissues of the embryo, mediated by aberrant activation of a PI3K-mTORC1-NFkappaB pathway (Hatzold et al., eLife 2016), thus constituting a novel embryonic animal model for carcinogenesis AND metastasis. Atp1b1a normally acts both as an osmo- and an epithelial polarity regulator, while the malignancy, but not the epithelial polarity defects, of basal keratinocytes of atp1b1a mutants is rescued upon isotonic treatment of the embryos. Together, this points to Atp1b1a as a novel tumor suppressor and to hypotonic stress as a novel tumor risk factor. Interestingly, similar to hai1a mutants, carcinogenesis in atp1b1a mutants is strictly dependent on the transmembrane cell surface protease ST14, which in this case is hyperactive because of its aberrant localization at the cell surface (resulting from the epithelial polarity defects) and its increased enzymatic activity (resulting from the hypotonic conditions). This indicates that ST14 can become hyperactivated by different means, leading to the activation of different signal transduction pathways and different cellular and oncogenic responses (hyperplasis both upon loss of Atp1b1a or Hai1a, metastasis only upon loss of ATP1b1a, but not upon loss of Hai1a). Comparative single cell RNA sequencing approaches are currently undertaken to further elucidate the differential mechanisms underlying these fundamentally different responses.
In addition, our findings obtained for zebrafish embryos are currently validated in various other cancer models, such as transgenic zebrafish with RAS-dependent carcinogenesis during adulthood (unpublished), as well as different human cancer cells lines (squamous cell carcinomas of the skin, prostate cancer, non-small cell lung cancer / NSCLC). In addition, systematic analyses of atp1b1a mutants are ongoing to identify the relevant (oncogenic) NFkappaB targets, as well as the crucial mediators activating the oncogenic PI3K-mTORC1-NFkappaB pathway in response to hypotonic stress and compromised epithelial polarity. Furthermore, chemical compound screens are applied to identify potential anti-carcinogenic drugs that rescue the malignancy in hai1 and/or atp1b1a mutants. Identified drugs will be subsequently tested, either singly or in combination, in adult zebrafish and mouse or human cancer cell models, with the ultimate goal to design improved anti-carcinogenic therapies.
Adult epidermis
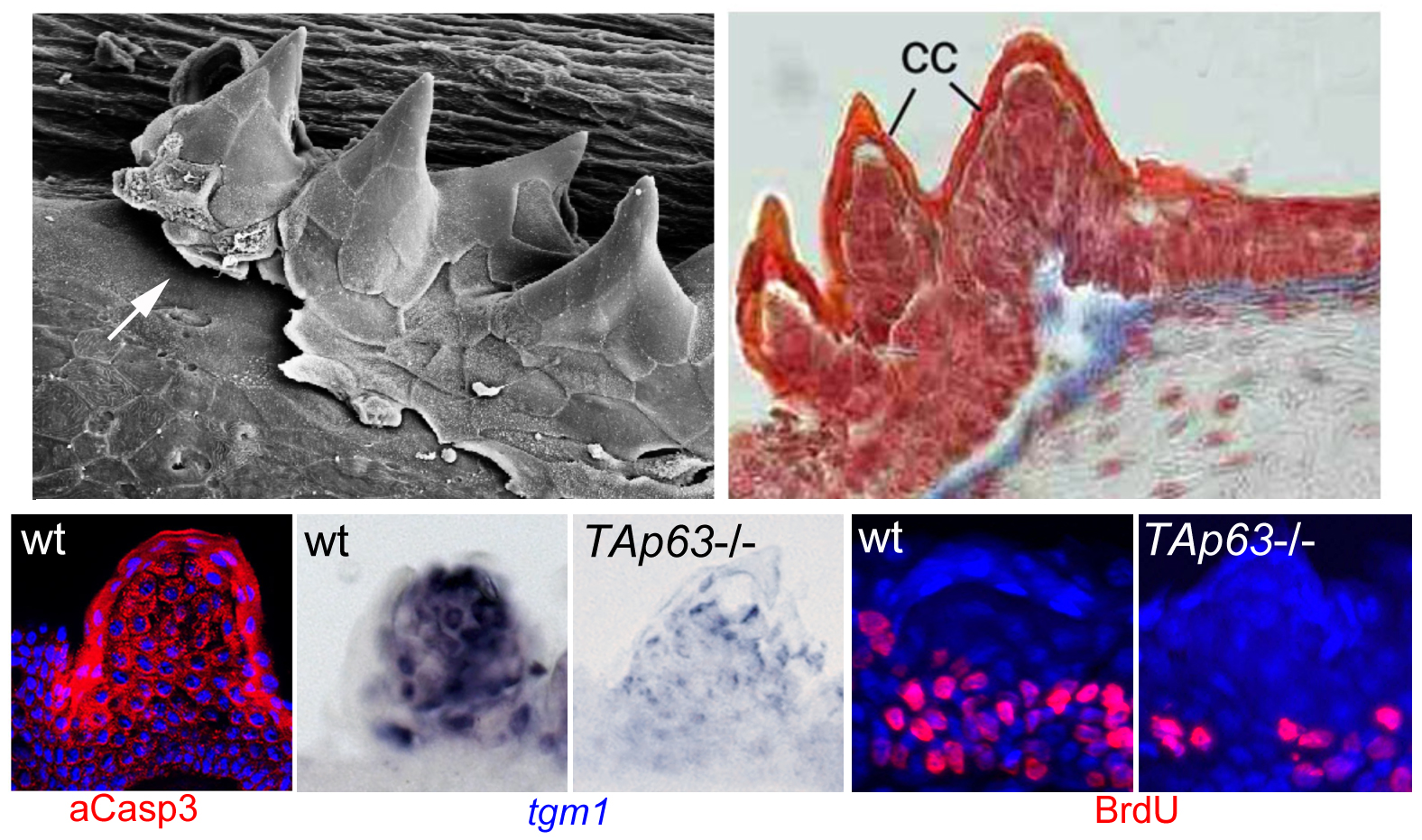
To follow up our studies on the role of p63 in the epidermis, we used a reverse genetics approach to generate zebrafish mutants specifically lacking the TAp63 isoform. In contrast to the ∆Np63 isoform (Bakkers et al., Dev Cell 2002; see above), the TAp63 isoform contains an N-terminal transcriptional transactivation domain, making it very similar to the well-known tumor suppressor p53. Recently, an N-terminally truncated isoform has also been identified for mammalian and fish p53, designated ∆113p53, which similar to the correlation between ∆Np63 and TAp63 largely acts as an antagonist of full length (TA)p53. Interestingly, in contrast to the loss of ∆Np63 function, loss of TAp63 does not affect early zebrafish skin development. Rather, mutants display specific defects in the so-called breeding tubercles, epidermal appendages of adult fish. According to our analyses, keratinocytes in these breeding tubercles undergo more advanced terminal differentiation than in the regular body epidermis, including cornification-like processes, cell desquamation at the surface and continuous self-renewal (Fischer et al., PLoS Genet. 2014), making the breeding tubercles a valuable model for the fully stratified epidermis of adult mammals / humans. In these breeding tubercles, ∆Np63 is restricted to basal-most keratinocytes (which serve as stem cells), while TAp63 is only active in upper / differentiating keratinocytes, activating a linear pathway involving Notch signaling and activation of caspase-3, a protease promoting cell death. TAp63 mutants, as well as fish treated with Notch or caspase-3 inhibitors, display compromised terminal differentiation of upper keratinocytes, as well as compromised proliferation of basal keratinocytes. Thus, by combining cell autonomous with non-cell autonomous effects, one and the same pathway seems to promote the loss and the replacement of cells, thereby ensuring proper tissue homeostasis and self-renewal. Defects are further enhanced in TAp63/p53 double mutants, pointing to thus far largely overseen partially redundant roles of these two related transcription factors (Fischer et al., PLoS Genet. 2014). Using transcriptomics and proteomics in addition to candidate testing approaches, we are currently searching for the relevant TAp63/p53, Notch or caspase-3 targets and substrates mediating keratinocyte differentiation versus keratinocyte differentiation. Furthermore, we have started to compare zebrafish mutants specifically lacking the ∆113p53, the TAp53 or both isoforms, both in the context of epidermal homeostasis in breeding tubercles and in the context of carcinogenesis. Finally, further zebrafish mutants from forward genetic screens with compromised breeding tubercle formation are being analyzed. The causative DNA lesions have been identified, and we try to understand how the products of the affected new genes are mechanistically linked to the formerly identified regulatory p53/p63 – Notch – caspase3 pathway. Interestingly, human homologs of these genes are mutated in different inherited diseases with skin or skin appendages abnormalities. Therefore, results obtained in zebrafish should also be relevant for a better understanding of these human diseases.
In addition, we have started to analyze self-renewal in regular body epidermis of adult zebrafish. Strikingly, hydroxyurea-treatments to block cell proliferation and cell lineage tracing experiments have revealed that approximately 6% of epidermal cells are replaced during one day, resembling the self-renewal rate of the cornified human epidermis. However, in contrast to cornification and desquamation that occurs in human and all land-based mammals, upper keratinocytes of the zebrafish seem to be lost via apical live cell extrusions (unpublished). Future research has to reveal how under these circumstances (absence of apoptosis) cell loss at the tissue surface is linked to cell birth in basal domains to assure tissue homeostasis.
Skin regeneration / wound healing
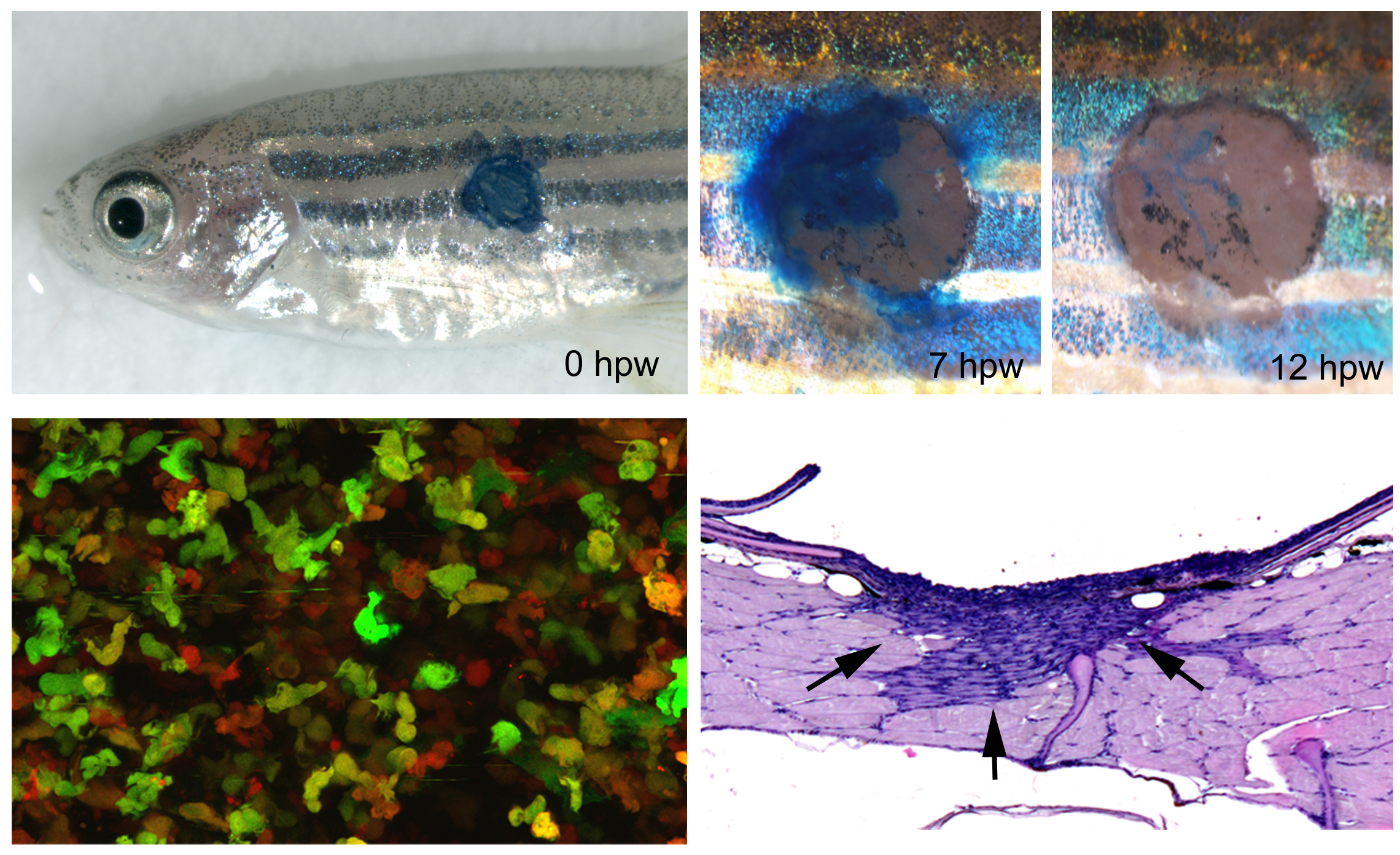
In addition to skin development, homeostasis and carcinogenesis, we study skin regeneration / cutaneous wound healing in adult zebrafish, applying both full-thickness wounds down to the level of the subcutaneous adipocytes and partial thickness wounds down to the level of the dermis. Pioneering studies revealed that apart from external clot formation, all steps of mammalian wound healing also take place in full-thickness wounds of adult zebrafish, such as re-epithelialization, inflammation by innate immune cells, granulation tissue formation by dermal fibroblasts, neo-vascularization, and tissue remodeling. However, compared to mammals, these different steps occur in a more consecutive manner, allowing a better dissection of primary and secondary effects caused by experimental interventions. In combination with pharmacological treatments and transgenic approaches for temporally controlled blockage of growth factor signaling, this allowed us to show that inflammation is a prerequisite for granulation tissue formation, with a stimulatory effect of Fibroblast Growth Factor (FGF) signaling from innate immune cells to dermal fibroblasts (Richardson et al., J Invest Dermatol 2013). In contrast, FGF signaling is dispensable for wound re-epithelialization, which instead depends on TGFbeta signaling to promote lamellipodial crawling of keratinocytes at the leading edge of the re-epithelializing epidermal tongue (“pulling” at the front; observed via live imaging of GFP-labeled keratinocytes of wounded fish!), and on progressive Rho/myosin-dependent long-range epithelial rearrangement such as cell flattening and radial cell intercalation in more remote regions of the epidermis (“pushing” from the back). These long-range re-arrangements assure keratinocyte recruitment from the adjacent epidermis into the wound bed and make re-epithelialization completely independent of keratinocyte proliferation, which is only required, stimulated by FGF signaling, for the later re-growth of the adjacent epidermis to its initial thickness (Richardson et al., Development 2016). Thereby, the zebrafish system allows to easily dissect mitogenic from motogenic effects of genetic insults or applied drugs, another experimental advantage compared to studies of wound healing in the mouse. Currently, using similar transgenic and pharmacological approaches, the roles of additional growth factors during the different steps of wound healing in adult zebrafish are studied.
Another crucial difference between cutaneous wound healing in zebrafish and mammals / human is that granulation tissue remodeling and resolution in the zebrafish is perfect, leaving no scar behind. Together with Prof. Sabine Eming from the Dermatology of the Cologne University Hospital, we have started to investigate the molecular and cellular basis for this crucial difference, using genetic manipulations to make wounds in the mouse heal in a scar-free manner like in zebrafish, and to make - in reverse - wounds in the zebrafish display a long-lasting fibrotic response as in mammals. First data point to the differential involvement of resistin-like molecules (whose genes are missing in fish genomes), which in the mouse are secreted by wound macrophages, stimulating the expression of lysylhydroxylase 2 (encoded by plod2 gene) in fibroblasts and thereby the formation of more resistant collagen crosslinks (Knipper et al., Immunity 2015). Accordingly, temporally controlled transgenic plod2 overexpression in zebrafish leads to significantly thicker collagen fibers and a slight increase in the size of the granulation tissue. This, however, does not compromise the latter granulation tissue resolution, indicating that other or additional mechanisms in addition to the Resistin-like molecule – Lysylhydroxylase 2 – axis must be at play to allow scar-free skin regeneration in fish (unpublished).